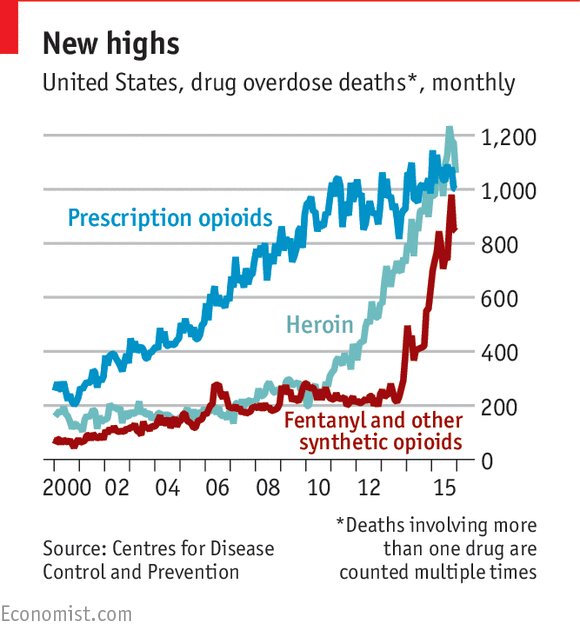
The opioid epidemic is thought to be due in part to
the over-prescription of drugs like OxyContin to treat patients with symptoms
of chronic pain. These drugs are known as a class as "opioids"
that all act via their interactions with the opioid receptors. By
understanding how these drugs work, we can develop new drugs that can still be
effective in treating pain, but lack the addictive properties of our currently
used drugs. We can also learn more about pain and chronic pain to develop
drug-free strategies that lessen the symptoms of pain.
How
do Opioids cause their effects? Opioids target opioid receptors
within the body in a variety of locations, the central nervous system,
peripheral nervous system as well as the gastrointestinal tract. The three
original major subtypes of opioid receptors are mu, kappa, and delta opioid
receptors within the body. The mu opioid receptor has three subtypes, µ1, µ2,
µ3 and is expressed in the brain spinal cord, intestinal tract, and sensory
neurons. The kappa opioid receptor also has three receptor subtypes, κ1 κ2 κ3,
and is expressed in the brain and the spinal cord. The delta opioid δ1 δ2
receptor is expressed in the brain and sensory neurons. Opioid receptors are G
protein coupled receptors that use opioids as their ligands and form hetero and
homotrimeric complexes that signal to kinase cascades and aid in the integrity
of a variety of proteins. Once the opioid is bound to the active site the
receptor undergoes a conformational change, which activates the intracellular G
proteins. The α subunit of the G-protein exchanges its bound GDP molecule for
an intracellular GTP molecule. This allows the α-GTP complex to dissociate from
the βγ complex. These complexes are now able to interact with target proteins.
Typically the opioid agonist binds to its G-protein receptor, which results in
the inhibition of adenylyl cyclase. This causes a reduction in intracellular
cyclic adenosine monophosphate (cAMP) levels. These complexes also interact
with a number of ion channels, producing activation of potassium and inhibition
of calcium. The net effect of these occurrences cause a reduction of
neurotransmitter release, which decreases the generation of the postsynaptic
impulse which inhibits tonic neuronal activity. Opioid receptors can also be
activated by endogenous peptides that are released by neurons in the brain.
There are three pro-hormone precursors that provide the parent compounds these
endogenous ligands. Proenkephalin, is cleaved to form met-enkephalin and
leu-enkephalin, and bind to the DOP (delta opioid receptor). Prodynorphin which
is cleaved to form Dynorphin A and B, these are agonists at the KOP (kappa
opioid receptor). β- endorphin is cleaved to form Pro-opiomelancortin (POMC),
which is an agonist at the MOP (mu opioid receptor). However, POMC is capable
of displaying agonist activity at all three classical opioid receptors.
Schumacher
MA, Basbaum AI, Naidu RK. Opioid Agonists & Antagonists. In: Katzung BG,
Trevor AJ. eds. Basic & Clinical Pharmacology, 13e New York, NY:
McGraw-Hill; 2015.
Sobczak, Marta, et al. “Physiology, Signaling, and
Pharmacology of Opioid Receptors and Their Ligands in the Gastrointestinal
Tract: Current Concepts and Future Perspectives.”NCBI, US National Library of Medicine, Feb. 2013,
www.ncbi.nlm.nih.gov/pmc/articles/PMC3895212/.
Biased agonism and tolerance. Opiods exert other actions that proceed via
events known as "biased agonism" and "tolerance".
These two events are related. Biased agonism occurs when a receptor has
affinity for multiple ligands. Depending on the ligand, a different signal
transduction pathway can be activated, but the ligand specifies which will be
activated. Tolerance is a reduced reaction to a drug after repeated use. When a
patient is prescribed continuous use of an opiate, tolerance can become a
problem in a clinical setting. Since opioids cause analgesic side effects, the
dose may be increased at times when the analgesic side effects are no longer as
strong. Opioid tolerance associated with biased agonism is currently not a
completely understood process, but ongoing research aims to understand the
effects. Some scientists argue that an opioid such as morphine may bind a
receptor, and even though the morphine is never internalized, the cell is still
signaling and causes a tolerance buildup to occur. Other ligands in the body
have the ability to become phosphorylated, which changes their structure, and
then are internalized and desensitized in the cell, which leads to tolerance.
Structurally different agonists for a receptor such as mu can produce different
binding efficiencies, which can influence the rate of arrestin recruitment or G
protein activation. Research is still being conducted in order to obtain more
clear results. Many researchers also hypothesize that because mu receptors
are recycled, tolerance is increased.
Williams, John T. et al. “Regulation of µ-Opioid Receptors: Desensitization,
Phosphorylation, Internalization, and Tolerance.” Ed. Annette C. Dolphin. Pharmacological Reviews 65.1 (2013):
223–254. PMC. Web. 18 Sept.
2017.
Al-Hasani, Ream, and Michael R. Bruchas. “Molecular Mechanisms
of Opioid Receptor-Dependent Signaling and Behavior.” Anesthesiology 115.6 (2011): 1363–1381. PMC. Web. 18 Sept. 2017.
Anderson, B.
(2011). A Pharmacological Primer of Biased Agonism. Endocrine, Metabolic and
Immune Disorders: Drug Targets.
doi:10.2174/187153011795564179
Other approaches that can be used to treat
pain. Drugs that can be administered include non steroidal anti-inflammatories, such as acetaminophen,
antidepressants, anticonvulsants, muscle relaxants, the use of nerve block
procedures (epidural steroid injections; facet joint injections; lumbar
sympathetic block) and natural alternatives (arnica gel; aromatherapy;
vitamin C). These drugs and approaches vary with respect to their
effectiveness and limitations. Nonsteroidal anti-inflammatory drugs,
like ibuprofen, can be a convenient and effective
Muscle relaxants can also be used to treat some kinds
of muscle pains,
Natural alternative are certainly an option and should
be considered (the less medication we have to take, the better). They may work
surprisingly well for mild pain, but these alternatives are likely to be
limited in their effectiveness in treating severe pain. Arnica gel can be used
for bruising and muscle soreness. Some users have said the arnica gel helped
control their arthritis on a level comparable to ibuprofen. Sufferers of
chronic pain like fibromyalgia could consider aromatherapy- for example,
massaging essential oils into areas of discomfort. Lastly, it is thought that
taking vitamin C supplements may sooth sufferers of some chronic pain, like
osteoarthritis.
Wolkerstorfer, A., Handler, N., & Buschmann, H. (2016).
New approaches to treating pain. Bioorganic & Medicinal
Chemistry Letters,26(4),
1103-1119. Retrieved September 18, 2017, from http://www.sciencedirect.com/science/article/pii/S0960894X15304169
Pain, R. (2000). Limitations of NSAIDs for pain management:
Toxicity or lack of efficacy? The Journal of Pain,1(3), 14-18. Retrieved September 18, 2017, from http://www.jpain.org/article/S1526-5900(00)09397-4/fulltext
Dean, L. (2011). Comparing NSAIDs. Pubmed: Clinical
Q&A. Retrieved
September 18, 2017, from https://www.ncbi.nlm.nih.gov/books/NBK45590/.
Munir, M., MD, Enany, N., MD, & Zhang, J., MD. (2007).
Nonopioid Analgesics. Medical Clinics of North America,91(1), 97-111. Retrieved September
18, 2017, from http://www.sciencedirect.com/science/article/pii/S0025712506001179?via%3Dihub
Lukatz, T. (2000). Anticonvulsants for neuropathic pain
syndromes: mechanisms of action and place in therapy. Drugs,60(5). Retrieved September 18, 2017,
from https://www.ncbi.nlm.nih.gov/pubmed/11129121.
Wiffen, P. (2013). Antiepileptic drugs to treat neuropathic
pain or fibromyalgia‐ an overview of Cochrane
reviews. Cochrane.
doi:10.1002/14651858.CD010567.pub2
Development of new drugs that target the opioid
receptor. Researchers at Tulane University have
developed a new painkiller drug that has a comparable strength to morphine but
has fewer side effects and likelihood of addiction. Here, several different endomorphin analogs,
referred to as EM analogs, were developed using an endogenous cyclized δ–amino
acid-containing endomorphin analog. Of the four different synthesized EM
analogs, the study found EM analog 4 to be the most effective. The engineered
EM analogs are highly selective for µ receptors (MOPs), which are the most
effective opioid analgesic targets. The EM analogs acted through binding
to the µ receptor, and further decreasing glial P38/CGRP/P2X7 receptor
signaling. Activation of this receptor signaling pathway has implications in
causing chronic pain.
The study was
performed as follows. First four
different EM analogs were synthesized.
Then, the activation of MOP, DOP,
KOP was ensured through completing receptor binding assays that utilized cloned
CHO-K1 membranes as the membrane for the respective receptors to function
within. To ensure the activation of these receptors by the EM analogs,
activation of the receptors was measured through completion of GTPgS assays,
which demonstrated over a 95% efficacy for successful MOP activation. Once it
was established that the opioid receptors were activated with the EM analogs,
trials progressed to an in vivo setting.
This study used both male CD-1 mice and Sprague-Dawley
rats that were exposed to a 12-hour light photoperiod/12-hour dark photoperiod.
Drug administration for the Sprague-Dawley rats used an indwelling jugular vein
catheter, whereas drug administration for the CD-1 mice was done either through
SQ injections in the neck region or through oral routes using a gavage. The
same measures were completed for both rats/mice injected with morphine as well
as mice/rats injected with EM analogs.
Antinociception was
tested through the “tail-flick” test, where rodents were exposed to a heat
source at their tail. Reaction latency was recorded with a nine second cutoff
time to prevent possible tissue damage. The duration of antinociception for IV,
SQ, and oral administration of EM analog 4 were all higher than the respective
durations of antinociception for morphine administration.
Respiratory
depression was measured in non-anesthetized rats that were allowed free
movement within a plethysmography system. Minute ventilation was monitored over
a 20-minute time span before drug administration, then again ever 20 minutes
after drug administration until antinociception levels had decreased to
>50%. Compared to morphine, rodents who were exposed to EM analogs had a
decreased level of respiratory depression, and even complete absence of
respiratory depression in some rodents.
Motor coordination
was examined using a rotorod system, which is a system that measures the
ability of an animal to remain upright while on a moving rod. Rodents were
given their respective drugs every twenty minutes until a %MPE of greater than
90% was achieved. From there the rats were left on the rotorods for 3 minutes
until being removed. Motor coordination was calculated using the Rotomex-5
instrument. Compared to rodents exposed to morphine, rodents who were exposed
to EM analogs did not have statistically significant motor coordination
impairment whereas their morphine rodent counterparts did.
Both hyperalgesia and
tolerance were tested over a seven-day period, and ED50s values
(effective dose values) were compared before and after the seven-day period.
Rodents exposed to EM analogs demonstrated less tolerance than the morphine
exposed rodents after a seven-day period. EM analog 4, when compared with
morphine, demonstrated no induced hyperalgesia whereas the rodents given
morphine showed induced hyperalgesia after the seven-day period.
The activation of
glial P38/CGRP/P2X7 receptor signaling was measured through using markers for
astrocytes, microglia, and MAP kinase and then examined for the expression of
the different markers within post-mortem rodents. The rodents injected with
morphine demonstrated a greater activation of cell receptor signaling than the
rodents injected with EM analogs, which indicates a reduced expression of
chronic pain within the EM analog rodents than the morphine rodents.
The risk of dependency
and abuse were measured in two different formats, both in conditioned place
preference settings and in self-administration settings. For the conditioned
place preference setting, rodents could freely roam in a two-compartment box
for two days. After two days, the rodents were given their respective opioid
drug and confined to only one of the compartments for a 30-minute duration.
This cycle of drug administration and 30-minute confinement was repeated for
three consecutive days. On the fourth day, the rodents were placed back in the
two-compartment box without being given their opioid drug and were allowed free
roaming between either compartment. The amount of time spent in each
compartment was recorded over a twenty-minute time span. The rodents that had been
injected with morphine spent a significantly greater amount of time in the
compartment in which they had been administered morphine in rather than the
other compartment. The rodents that had been injected with EM analog, however,
and demonstrated no preference between the two compartments. For the
self-administration setting, rodents were placed in a compartment that had an
“active” bar that when pushed on administered their respective drug
intravenously, and an “inactive” bar that when pressed did not cause drug
release. Infusion release was regulated to only allow a fixed ratio of drug
administration over a seven-day period with exposure to the different bars for
12 hours per day. The rodents that received morphine exhibited an increase
“active” bar pressing by a five-fold increase. The rodents that received EM
analog after pressing their “active” bar did not demonstrate a more frequent
bar pressing over the seven-day period.
One
of the limitations of this study was the limited comparison to morphine
exposure and not any of the other available opioid drugs. While morphine is a
commonly used opioid both in the medical profession and in illegal-use,
morphine is not the only drug used in both settings. This study also only
compared the effects of morphine and the EM analogs in two rodent species.
Another limitation of this study is the duration of time that these
measurements were taken over, which fail to demonstrate the same side effects
of their drug usage over a longer time interval. Many opioid drugs demonstrate
greater side effects and complications during prolonged use for chronic pain,
and an experimental setting that only examined these parameters concerning
opioid drug side effects during a seven-day period cannot be used to predict possible
implications on the same drug usage over a longer interval.
While
the EM analogs described in this study, particularly EM analog 4, demonstrated
an improved analgesic-to-side-effect ratio, several steps need to be taken to
determine the possible applications of EM analog 4 in the medical field.
Similar studies need to be done that compare other opioid drugs, such as
fentanyl and codeine. Similar studies also need to be completed over a larger
time span to determine the applications of EM analog 4 in the treatment of
chronic pain, seeing that many of the instances that use opioid drugs for the
management of chronic pain. Once studies such as these have been completed
within the rodent species, comparable experiments need to be conducted using
primate species before any human implications can be derived through the
substitution of EM analogs for pain management in the medical field.
Zadina, J., Nilges, M., Morgenweck, J., Zhang, X., Hacker,
L., & Fasold, M. (2015). Endomorphin analog analgesics with reduced abuse
liability, respiratory depression, motor impairment, tolerance, and glial
activation relative to morphine. Neuropharmacology,105, 215-227. Retrieved September 18,
2017, from http://www.sciencedirect.com/science/article/pii/S0028390815302203
No comments:
Post a Comment