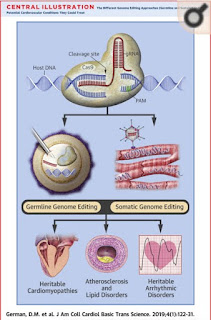
Red blood cell antigens are best known as A, B,
AB, and O, however, up to 342 identified independent antigens can be present on
a single red blood cell.1 The present antigens on an individual’s
red blood cells are recognized and processed by their immune system as
originating from themselves.5 When an antigen that is not typically
present is found by an immune cell, the immune system deems the red blood cell
as foreign and proceeds to attack it and others with the same antigen.5
For this reason, when an individual needs a blood transfusion, they must
receive their donation from someone who has compatible antigens on their red
blood cells.5 This can become difficult when the individual has a
more rare pattern of blood antigens on their blood cells or in other words, a
rare blood type.
 |
| Figure 1. The common blood types with their coinciding alloantibodies. |
When blood types such as A or AB are discussed,
they are mentioned in the context of if the individual is positive or negative
for them. When the individual is negative for the antigen, they can donate to
an individual that is positive or negative for the antigen but can only receive
a donation from an individual negative for it.5 The most discussed
blood types are A+, B+, AB+, O+, A-, B-, AB-, and O-.5 The most well
recognized flexible blood donor group is O- which is known as the universal
donor because they are able to donate to all previously mentioned blood types.5
The most widely compatible blood group is known as golden blood and individuals
with this extremely rare blood type are inherently able to donate to all rare
blood types from the Rh antigen system and even common blood types.6,11
Rh-deficiency syndrome, an autosomal recessive
genetic disorder characterized by red blood cells that lack all 61 antigens
known as Rh system antigens, can occur in both a complete (Rhnull) or
significantly reduced phenotype (Rhmod) .2,6 The syndrome is
extremely rare, it has been reported less than 50 times since its discovery in
1960.11 Other rare blood types from the Rh antigen system can lack
any one or more of the Rh antigens but less than all. The low prevalence of the
Rhnull phenotype and the potential complexity of the other Rh system blood
types makes finding a blood donor for a patient with these blood types extremely
hard. This is where the coined term ‘golden blood’ is derived. Afflicted
individuals are classified into two subgroups based on their specific genetic
defect; regulatory when the mutation is in a suppressor gene and amorph when it
is at the Rh gene locus.2,9 Patients with Rhnull syndrome are found
to have stomatocytes which are osmotically fragile red blood cells that undergo
hemolysis when in a hypotonic environment.2,3,8 This is a result of
their abnormal shape that seems to coincide with the lack of the cell membrane
antigens.3 As a result of this the patients also suffer from chronic
hemolytic anemia to some degree.2,3 This is a condition in which the
patients’ blood cells are broken down via hemolysis faster than they are made.7
Individuals with hemolytic anemia have symptoms such as fatigue, pale
skin, chills, fever, heart palpitations, confusion, and more.10 Clinically
Rh deficient patients readily produce alloantibodies when exposed to Rh
antigens and in certain situations such as pregnancy can be very dangerous.2,4
 |
| Figure 2. Stomatocytes in culture. |
When a Rhnull mother is pregnant with a fetus
with the Rh antigens (Rh+) the mother's immune system may create antibodies
against the fetus’s red blood cells and destroy them.1 This can
cause hemolytic anemia in the fetus and the severity of the anemia can induce
brain damage, severe illness, or even death of the fetus.1 Expecting
mothers whose red blood cells are lacking any of the Rh system antigens and
have not yet produced antibodies towards her growing fetus may be put on Rh
immunoglobulin (Rhlg) at around the 28th week of pregnancy.1
This can prevent the mother from starting to produce antibodies to the fetal
blood cells for the remainder of the pregnancy.1 The mother is also
given another dose of the Rhlg post-birth to prevent any Rh+ cells left in the
mother from producing an immunological reaction.1 Any Rh- mothers
that produced antibodies to Rh+ fetal blood will not be helped by Rhlg
treatment.1 While this condition may only affect a select few
expecting mothers however, it can make their pregnancy very difficult and
scary.
 |
| Figure 3. Illustration of fetal hemolytic disease development |
As previously mentioned, individuals with
golden blood are able to donate to anyone with common or specific rare Rh
system blood types without their blood generating an immune response in the
recipient.6 Before Rhnull blood was first discovered it was thought
an individual without these antigens would not survive utero.6 Since
its discovery, golden blood has been sought after by researchers and physicians
alike for its rare properties, however, in 2017 there were only 9 active
donors.11 This makes finding a single bag of the blood extremely
difficult which has made blood transfusions for these patients complicated and
slowed the progression of studies on the blood.11 Despite
this, with all of its very special characterizations, these individuals’ blood
holds an extraordinary place in the healthcare and scientific communities.6
References:
1. Rh Factor. American Pregnancy
Association. https://americanpregnancy.org/pregnancy-complications/rh-factor/.
Published October 9, 2019.
2. Journal Of Pakistan Medical
Association. JPMA. https://jpma.org.pk/article-details/2399.
3.
Stomatocyte. Stomatocyte - an
overview | ScienceDirect Topics.
https://www.sciencedirect.com/topics/medicine-and-dentistry/stomatocyte.
4.
Glossary: Alloantibody - Blood Bank
Guy Glossary. Blood Bank Guy. https://www.bbguy.org/education/glossary/gla17/.
5.
Blood: the basics. Professional
Education.
https://professionaleducation.blood.ca/en/transfusion/publications/blood-basics.
Published February 11, 2019.
6. Bailey P. Wellcome. The man with the
golden blood. https://mosaicscience.com/story/man-golden-blood/.
7.
Hemolytic Anemia. Johns Hopkins
Medicine.
https://www.hopkinsmedicine.org/health/conditions-and-diseases/hemolytic-anemia.
8.
Osmotic fragility test:
MedlinePlus Medical Encyclopedia. MedlinePlus.
https://medlineplus.gov/ency/article/003641.htm.
9. Cartron, Jean-Pierre.
"Rh-deficiency syndrome." The Lancet 358 (2001):
S57.
10.
Hemolytic Anemia. National Heart
Lung and Blood Institute.
https://www.nhlbi.nih.gov/health-topics/hemolytic-anemia.
11.
Rhnull, the Rarest Blood Type on
Earth, Has Been Called the "Golden Blood". Curiosity.com.
https://curiosity.com/topics/rhnull-the-rarest-blood-type-on-earth-has-been-called-the-golden-blood-curiosity/.
By Cheyenne Cook, A Master’s of Medical
Sciences Student at the University of Kentucky